Huntington’s disease (HD) is an inherited, progressive neurodegenerative disorder characterized by chorea, gradual but inexorable cognitive decline, and psychiatric disturbances. Patients usually die 10-15 years after HD onset, and there is no cure
for this devastating neurological disease. Despite extensive studies regarding the toxic function of mutant huntingtin protein (mHtt), the exact mechanism(s) behind the selective neuronal loss of striatal medium spiny neuron is still unknown. Oxidative
damage idea has long been suggested to be important in pathology of many neurodegenerative diseases such as Parkinson’s and HD, however, the origin of the oxidant and the reason why specific brain region is under higher level of oxidative damage in
a particular disease remain obscure.
Mitochondrion is not only a very efficient Ca2+ buffer system, but also constitutes the major ROS generator in eukaryotic cells including neurons. A research group (led by Dr. Tang) from the Institute of Zoology, Chinese Academy of Sciences
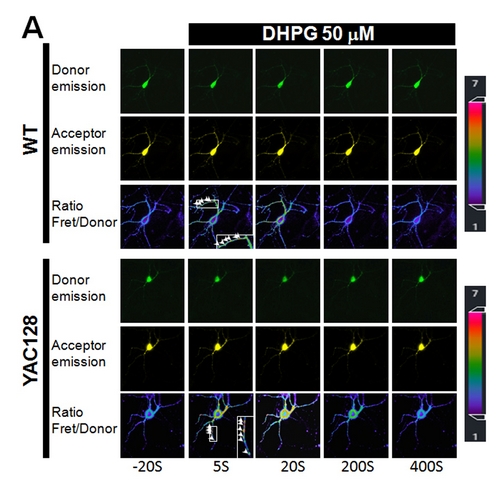
aimed to look at the potential connection between mitochondrial Ca2+ loading and superoxide generation in HD cells/neurons. By using a YAC128 HD mouse as a model system, and highly sensitive mitochondria-targeted superoxide indicator (cpYFP)
and matrix Ca2+ indicator (cameleon), researchers found that HD cellsexhibit a strikingly higher level of mitochondrial matrix Ca2+ loading and elevated superoxide generation compared to WT cells, and this higher level of mitochondrial
oxidant stress is critically dependent on mitochondrial Ca2+ loading. More importantly, mitochondrial Ca2+ loading in HD cells caused a 2-fold higher level of mitochondrial genomic DNA (mtDNA) damage due to the excessive oxidant
generation. These data demonstrated a new causal link between dysregulated mitochondrial Ca2+ signaling, elevated mitochondrial oxidant stress and mtDNA damage in HD, and shed a new light on the underlying mechanism by which mHtt protein
causes elevated oxidant stress in HD neurons. This work also point to a therapeutic strategy for HD, that is reducing mitochondrial Ca2+ uptake may effectively protect striatal neurons from death in HD.
Previous works (including Dr. Tang’s own works) demonstrated that mHtt protein preferentially augments intracellular Ca2+ signaling in striatal neurons in HD via NR2B, IP3R1 and D1-dopamine receptors, thus higher mitochondrial Ca2+ loading and elevated mitochondrial oxidant stress are expected to occur selectively in striatal region which may explain the brain regional specificity of oxidant stress.
“We believe that our present study provides authentic evidences to support a new link between deranged mitochondrial Ca2+ signaling, elevated mitochondrial oxidant stress and mtDNA damage in HD. Gradual accumulation of the detrimental oxidant
damage to mtDNA and mitochondrial proteins could cause the dysfunction of mitochondria and finally the neuronal death in HD”, said by Tie-Shan Tang, the corresponding author of the paper. “A combination of reducing mitochondrial Ca2+ uptake
and scavenging mitochondrial oxidant could form a therapeutic approach to slow the progression of HD, and we consider this information is important for scientists who are working on polyQ diseases and other neurodegenerative diseases such as Parkinson’s
and Alzheimer’s as well”, Dr. Tang added.
PhD student Jiuqiang Wang in Dr. Tang’s lab is the first author of this work. Other collaborators include Dr. Heping Chen (Peking Univ), Dr. Qinmiao Sun and Dr. Quan Chen (IOZ), and Dr. Caixia Guo (Beijing Institute of Genomics). This work was supported
by grants from the National Basic Research Program of China (2011CB965003, 2012CB944702, 2011CB809102), NSFC30970931, NSFC31130067 and by grant from the Knowledge Innovation Program of Chinese Academy of Sciences (KSCX2-YW-R-148).